18 November 2021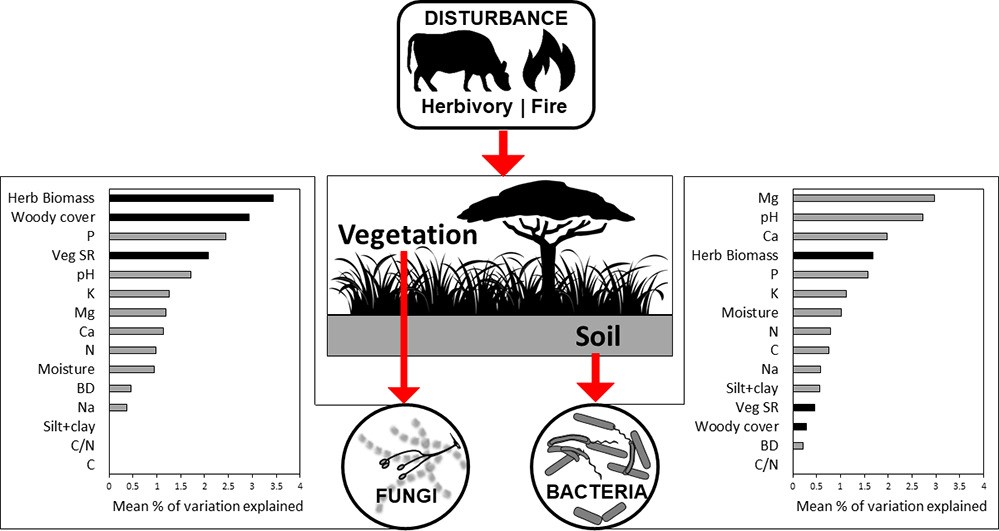 Fire and herbivory drive fungal and bacterial communities through distinct above- and belowground mechanisms Fire and herbivory drive fungal and bacterial communities through distinct above- and belowground mechanisms
Citation: Vermeire M-L, Thoresen J, Lennard K, Vikram S, Kirkman K, Swemmer AM, Te Beest M, Siebert F, Gordijn P, Venter Z, Brunel C, Wolfaard G, Krumins JA, Cramer MD, Hawkins H-J (2021). Fire and herbivory drive fungal and bacterial communities through distinct above- and belowground mechanisms. Science of the Total Environment, volume 785, (2021). doi.org/10.1016/j.scitotenv.2021.147189▼ Read summaryby Thato Motlhalamme, South African Grape and Wine Research Institute, Stellenbosch University, South Africa Grasslands and savannah are the most abundant vegetation globally, providing habitats for macro and microorganisms as well as a variety of ecosystem goods and services. Grasslands are home to many herbivores e.g., kudu and eland that graze the land. However, these habitats are also subjected to periodic fires (either wild or controlled). Both these factors influence the structure of grassy ecosystems and their microbial components. Microbial communities in the soil play an important role in breaking down organic matter, acquiring nutrients for plants and growth in these biomes. Although studies have looked at how fire and herbivory induce changes in vegetation and soil affect microbial communities, it is still not clear what the long-term effects of fire and herbivory are on microbial communities or how regional (e.g., biome) and local (e.g., riparian zone) contexts modify these effects. Vermiere et al characterised soil physico-chemical properties, plant species richness and biomass, microbial biomass and bacterial and fungal community composition and diversity in six long-term (18 to 70 years) ecological research sites in South African savanna and grassland ecosystems. The study found that soil physiochemical properties, plant species richness and standing biomass were significantly modified by the fire and herbivory regimes applied in the research sites. However, bacterial and fungal communities were shaped by different driving forces. Edaphic properties (Mg, pH, Ca) drove changes in bacteria whereas vegetation (herbaceous biomass and woody cover) impacted the fungal communities. Fire and herbivory explained on average 7.5 and 9.8% of the fungal community variability and 6.0 and 5.6% for bacteria, respectively. The impact of edaphic variables, particularly pH in shaping bacterial communities is well documented but this study highlighted the importance of exchangeable cations (Mg and Ca) in shaping this microbial community. These cations are some of the main ions associated with cation exchange capacity (CEC) which is a measure of the soil’s ability to hold positively charged ions. CEC plays an important role in influencing soil structure and stability, nutrient availability, soil pH soil reaction to fertilisers and ameliorants. This study found that both fire and herbivory induced changes in the available nutrient cations and were the main correlates of bacterial diversity after pH. The study authors acknowledged that the changes in microbial communities in the long-term ecological research sites selected were muted but this may be linked to the ability of the microorganisms to evolve/adapt to the disturbances introduced. To truly understand the impact of fire and herbivory regimes on microbial communities, especially in African landscapes, longer timescales should be investigated. It is a very important soil property influencing soil structure stability, nutrient availability, soil pH and the soil’s reaction to fertilisers and other ameliorants. About the first author: Marie-Liesse Vermeire is a soil biogeochemist and ecologist. Her past research activities looked at the role of silicon nutrition in decreasing plant disease impact (MSc) and mineral-biota interactions in stabilizing carbon in soils (PhD). Her current research focuses on the interactions within soil food webs: how do they respond to fire, herbivory and agricultural practices; management of soil biodiversity, especially microbial communities; and the use of soil organisms in sustaining agriculture? Currently Marie-Liesse is a researcher at the CIRAD research centre (French agricultural research and cooperation organization working for the sustainable development of tropical and Mediterranean regions). Her research unit, "Recycling and risk", works to propose solutions for recycling organic matter and waste (livestock effluents, biosolids from wastewater treatment, agro-industry effluents, green waste and household waste) through farming practices with controlled agro-environmental risks. She hopes to generate the basic knowledge required to design “biologically intensive” agriculture, relying on ecological interactions and soil processes in view of amplifying the natural services that soils can provide to develop this concept of agriculture. |
18 November 2021 Impact of land use on bacterial diversity and community structure in temperate pine and indigenous forest soils Impact of land use on bacterial diversity and community structure in temperate pine and indigenous forest soils
Citation: Amoo AE, Babalola OO. 2019. Impact of land use on bacterial diversity and community structure in temperate pine and indigenous forest soils. Diversity, 11(11), 217; https://doi.org/10.3390/d11110217.
▼ Read summaryby Ben Jesuorsemwen Enagbonma, University of Benin, Nigeria Since the work of microbiologists such as Robert Koch et al. more than 100 years ago, we’ve known that some bacteria cause diseases. We are now entering an exciting new era where we are beginning to recognize that some bacteria could be used to prevent or even cure diseases. Helpful bacteria employed in this way are referred to as ‘probiotics’, usually in the context of applying live microorganisms to improve human health. The rise in human stresses on the environment such as conversion of natural forests to plantation to get their own gain, remains a strong factor affecting the variety and variability of species. The understanding of the consequences of land use change on soil bacterial communities is indispensable as part of global efforts for ecosystem restoration. Soil bacteria function as key drivers of biochemical processes; thus; Amoo and Babalola assessed the impact of land use on bacterial diversity and community structure in temperate pine and indigenous forest soils by sequencing the 16S rRNA gene V4 variable region using a MiSeq sequencer (Illumina, Inc. San Diego, CA, USA). After pre-processing, analysis of the sequenced reads was carried out using the MR DNA analysis pipeline and MG-RAST pipeline. Operational taxonomic units (OTUs) were defined by clustering at 3% divergence (97% similarity). Thereafter, OTUs per sample were taxonomically categorized via BLASTn against a curated database derived from RDPII and NCBI. Just as predicted by Amoo and Babalola 2019, the bacterial diversity and composition between the indigenous forests in relation to temperate pine plantations were affected by land use change across the sites with relatively higher bacterial richness and diversity in the indigenous forests in relation to the temperate pine forests. Amoo and Babalola investigation held variations in forest management practices and environmental variables such as soil physical and chemical properties as the vital drivers of forest soil bacterial diversity, abundance and community composition.  About the first author: Amoo Adenike Eunice received her PhD in Science Biology from North West University, South Africa in April 2018. At North West University, she participated in several research tours to South African Forests precisely located in Tweefontein and Witklip. After her doctoral studies, she went back to her Job in Nigeria, on a short-term visit as a Lecturer in Bowen University, however, her scholastic performances during her PhD program earned her a Postdoc position in Microbial Biotech Lab (South Africa). About the first author: Amoo Adenike Eunice received her PhD in Science Biology from North West University, South Africa in April 2018. At North West University, she participated in several research tours to South African Forests precisely located in Tweefontein and Witklip. After her doctoral studies, she went back to her Job in Nigeria, on a short-term visit as a Lecturer in Bowen University, however, her scholastic performances during her PhD program earned her a Postdoc position in Microbial Biotech Lab (South Africa).
|
23 October 2021A rooted phylogeny resolves early bacterial evolutionCitation: Coleman GA, Davín AA, Mahendrarajah TA, Szánthó LL, Spang A, Hugenholtz P, Szöllősi GJ, Williams TA. A rooted phylogeny resolves early bacterial evolution. Science. 2021 May 7;372(6542):eabe0511. doi: 10.1126/science.abe0511. PMID: 33958449.▼ Read summaryby Zichao Zeng, Yinzhao Wang There are two major types of cells: prokaryotes and eukaryotes. The difference between them lies in the nuclear structure and evolution level. The former includes Bacteria and Archaea, while the latter includes fungi, protozoa, algae, higher plants and animals. One hypothesis considers Eukaryotic cells were originated from the symbiosis of one bacterial cell into one archaeal cell. But how did the very first bacteria and archaea look like? This problem has been lingering in scientists’ minds for a long time. Although there is already a huge amount of data related to bacterial and archaeal genomes, it is still challenging to determine their Last Common Ancestors (e.g. the last bacterial common ancestor, LBCA) and the root of the archaeal and bacterial phylogenetic tree. A recent phylogenetic analysis carried out in collaboration between the University of Bristol, Eötvös University, Netherlands Institute for Sea Research and University of Queensland, published in Science, suggested that the root of the bacterial tree should be placed between Gracilicutes and Terrabacteria, which are two major bacterial clades, rather than between the Candidate Phyla Radiation (CPR) and all other Bacteria as suggested previously. To reduce the influence of long branch attraction and the uncertainty of the position of the universal root, Gareth A. Coleman et al. reconstructed and rooted the bacterial tree without an archaeal outgroup by applying a hierarchical phylogenomic approach. Horizontal gene transfer (HGT) is ubiquitous in bacteria, and standard models consider HGT as an obstacle that needs to be removed during the reconstruction of the tree of life. However, using gene tree-species tree reconciliation method to unite tree and metabolic network-based approaches, the authors modeled both the vertical and horizontal components of bacterial evolution. They found that horizontal and vertical evolutions are complementary to each other. Both contribute to a better understanding of bacterial phylogeny. Then they inferred that the Last Bacterial Common Ancestor was a rod-shaped double-membraned cell capable of motility, chemotaxis and with a type III CRISPR-Cas system. “Where do we come from? What are we? Where are we going?” Today, we are still pondering the answers to these philosophical questions. There is still a lot work to do in order to gain a comprehensive knowledge of the origin and early evolutionary history of life on our home planet Earth.
|
23 October 2021A standardized archaeal taxonomy for the Genome Taxonomy DatabaseCitation: Rinke C, Chuvochina M, Mussig AJ, Chaumeil PA, Davín AA, Waite DW, Whitman WB, Parks DH, Hugenholtz P. A standardized archaeal taxonomy for the Genome Taxonomy Database. Nature Microbiology 6, 946–959 (2021). doi.org/10.1038/s41564-021-00918-8.▼ Read summaryby Hungchia Huang, Yinzhao Wang Archaea are one of the primary divisions of life alongside Bacteria and Eukarya, and are considered to be an ancient form of life on Earth. Recently, the origin of Eukarya from within Archaea has been proposed, suggesting a two domain tree of life. Archaea are also known to play pivotal roles in elemental cycling in nature. However, despite these many exciting findings of Archaea in recent years, obtained mostly by metagenomic sequencing and analysis, Archaea are still full of mystery to scientific communities in both Earth and Life Sciences, and their phylogenetic relationships have not been systemically summarized. Hence, it is essential to collect genomic data to develop systematic, normalized taxonomies based on evolutionary relationships. The Genome Taxonomy Database (GTDB) has established a revised bacterial taxonomy, and a recent addition to the GTDB published by Dr. Christian Rinke at The University of Queensland in Nature Microbiology proposed a new standardized archaeal taxonomy including 2,392 archaeal genomes. The taxonomy is based on a phylogenetic framework of 122 concatenated proteins, resolves polyphyletic groups and normalizes ranks based on relative evolutionary divergence. The authors reclassified the archaeal taxonomy from four superphyla, including more than 30 phyla in total, to 16 archaeal phyla. The major changes are the reclassification of 3 main monophyletic units in the former Euryarchaeota to phylum-level lineages; unification of the Thaumarchaeota, Aigarchaeota, Crenarchaeota, Korarchaeota (TACK) superphylum into a single phylum; and uniting all Asgard phyla, which form a monophyletic group previously considered to represent a superphylum, into a single phylum. This study demonstrates that the proposed archaeal taxonomy effectively resolves the widespread incomplete and uneven classifications, as demonstrated by comparing the rank-normalized archaeal GTDB taxonomy with NCBI taxonomy. The authors also show that the taxonomy is robust against a series of phylogenetic variables, including marker gene selection, inference methods, corrections for rate heterogeneity or compositional bias, tree rooting scenarios and expansion of the genome database. In addition, the authors emphasized that it remains questionable whether GTDB defined bacterial phyla and archaeal phyla are comparable, as each taxonomy was developed independently. Overall, this standardized archaeal taxonomy is user-friendly, provides a complete taxonomy from domain to species, and allows to identify and classify novel genomes. User feedback is encouraged via the online forum on the GTDB website. The authors believe that the archaeal taxonomy will expand rapidly due to the increasing rate of genome depositions in the public repositories. |
29 September 2021Benchmarking microbial growth rate predictions from metagenomesCitation: Long AM, Hou S, Ignacio-Espinoza JC, Fuhrman JA. (2021). Benchmarking microbial growth rate predictions from metagenomes. The ISME Journal 15:183–195.▼ Read summaryI regularly remind myself and my students that you will always come up with an answer when you apply some analysis to your data, but how do you know whether the answer you get is right? This can be particularly challenging when studying uncultured microorganisms in the environment because it isn't always clear what the right controls are. Work by Long and his colleagues sought to answer this question for techniques that have become increasingly popular to use to measure microbial growth characteristics of uncultured microorganisms using metagenomic data. Measurements or estimates of microbial growth can reveal the important role these tiny creatures play in the ecosystem. Yet, a majority of environmental microorganisms can't be cultured to determine their maximum growth rate or identify how changing environmental conditions influences their growth. Even for organisms in culture, it is difficult to determine the actual growth rates that microorganisms experience under in situ environmental conditions because environmental conditions are difficult to recreate in the laboratory. Thus, techniques to estimate these parameters from metagenomic data have been developed, either through inherent genome characteristics (i.e. codon usage bias, CUB) or the distribution of reads or observations over the genome (e.g. peak-to-trough ratio, PTR). While these techniques were tested and validated with cultured microorganisms, the effectiveness of these techniques for uncultured microorganisms using shotgun metagenomics had not been thoroughly validated. The work by Long and colleagues more rigorously tests the ability of a variety of techniques to estimate these growth characteristics using methodology typical for environmental studies. By applying these techniques within experimental incubations of natural seawater, filtered to remove grazers and viruses so that changes in cell number better approximate growth, they compared the observed growth to growth estimated by CUB or PTR with metagenomics. CUB estimates were significantly correlated with maximum growth, suggesting they are a reasonable measure of the maximum growth rate for a diverse range of environmental marine microbes. However, PTR estimates were not well correlated to growth for the entire community and only showed positive correlations with growth for a subset of the community. Interestingly, Gammaproteobacteria was one taxonomic group for which this technique worked well. Thus, this work better defines the limitations of these approaches and highlights the importance of continuing to ask this basic question of your analysis- how do you know whether the answer you get is right? |
29 September 2021Depth-discrete metagenomics reveals the roles of microbes in biogeochemical cycling in the tropical freshwater Lake TanganyikaCitation: Tran PQ, Bachand SC, McIntyre PB, Kraemer BM, Vadeboncoeur Y, Kimirei IA, Tamatamah R, McMahon KD, Anantharaman K. (2021) Depth-discrete metagenomics reveals the roles of microbes in biogeochemical cycling in the tropical freshwater Lake Tanganyika. The ISME Journal 15:1971–1986.▼ Read summaryLake Tanganyika is a unique ecosystem in many ways and the work by Tran and colleagues provides an in-depth analysis into the diversity and function of the microorganisms that inhabit it. Lake Tanganyika is the second largest freshwater by volume in the world, harboring 16% of the Earth's freshwater. Microbial processes within this ecosystem have a major impact on local biogeochemical cycles, such as the generation and consumption of substantial amounts of methane (23 Tg) that collect below the depth where oxygen concentrations become undetectable. Freshwater tropical lakes are typically nitrogen-limited, so nitrogen cycling can influence the food web and overall productivity of the lake. By conducting shotgun metagenomic sequencing of 24 samples from depths up to 1200 m and metagenomic binning to reconstruct 523 metagenome assembled genomes, the authors were able to identify a number of unique and unexpected microorganisms and their associated biogeochemical functions. Among their discoveries is a novel species (Candidatus Tanganyikabacteria) most closely related to the non-photosynthetic Cyanobacteria-like Sericytochromatia. Sericytochromatia genomes had not been reconstructed and functionally described from the water column of freshwater lakes before. They also found that Lake Tanganyika had a large number of unique reconstructed genomes compared to those found in the deepest freshwater lake in the world, Lake Baikal. The microorganisms unique to Lake Tanganyika are likely related to the differences in geochemical parameters between the lakes, as Lake Baikal is oxygenated at depth and is seasonally ice-covered. Nitrogen-fixation was not found in any Cyanobacterial genomes, but within a diversity of organisms that were more similar to those of a stratified, humic lake. Denitrification potential was found in reconstructed genomes at a similar relative abundance at all depths. Bacteria capable of anammox, a process that also result in a loss of fixed nitrogen from this system, were also found throughout the lake. As climate change is expected to increase the number of stratified, anoxic lakes globally, this study provides an insight into the distribution of biogeochemical functions mediated by microorganisms that have important ecosystem functions within these systems. |
30 August 2021  Bacterial controlled mitigation of dysbiosis in a seaweed disease Citation: Li J, Majzoub ME, Marzinelli EM, Dai Z, Thomas T, & Egan S. (2021). Bacterial controlled mitigation of dysbiosis in a seaweed disease. ISME J (2021). https://doi.org/10.1038/s41396-021-01070-1.▼ Read summarySince the work of microbiologists such as Robert Koch et al. more than 100 years ago, we’ve known that some bacteria cause diseases. We are now entering an exciting new era where we are beginning to recognize that some bacteria could be used to prevent or even cure diseases. Helpful bacteria employed in this way are referred to as ‘probiotics’, usually in the context of applying live microorganisms to improve human health. A recent study by Jiasui Li et al, in a team led by Suhelen Egan at the Centre for Marine Science and Innovation, University of New South Wales, contributes to the emerging field of probiotics for application in environments such as the oceans, to improve or maintain marine organism health. The disease in question is bleaching of the red macroalga Delisea pulchra, which causes the algae to lose photosynthetic pigments. Like numerous diseases of marine organisms, D. pulchra bleaching is associated with the presence of particular bacteria that can trigger the disease, in this case some Phaeobacter, Aquinimarina, Agarivorans, and Alteromonas species. The team used a diffusion-based plate agar bioassay to assess the potential of hundreds of strains isolated from healthy seaweeds to inhibit growth of these potential pathogens. One strain was of particular interest – Phaeobacter sp. BS5 (not to be confused with the disease-triggering Phaeobacter strains!), which inhibited the growth of several pathogens. Likewise, adding Phaeobacter sp. BS5 to seaweeds in aquaria had a significant protective effect when the pathogen Aquinimarina sp. AD1 was also added. Considering the inhibitory growth effects of Phaeobacter sp. BS5 on the pathogens in plate assays, the team expected a negative correlation between the relative abundance of these two organisms on the experimental seaweeds – indicating that the protective strain directly inhibits pathogen growth on the seaweeds as it did in the lab. Surprisingly, this was not the case. Instead, 16S rRNA gene amplicon assays revealed a general shift in microbial community composition in the presence of the pathogen, which did not take place when pathogen and Phaeobacter BS5 were both applied. The protective effect might therefore be due to indirect interactions, for example, through Phaeobacter sp. BS5 strengthening the resistance of the native microbiome to disturbance, rather than direct interactions between probiotic and pathogen. This result is intriguing because it expands the range of microbes that could potentially be harnessed as probiotics. Even strains that do not directly affect pathogen growth could still protect against disease through interactions with other members of the native microbiome. This study also provides another fascinating example of how even closely-related microbial strains have the potential to cause or prevent disease, which are arguably diabolically opposite functional roles. The taxonomic groups studied here including Phaeobacter, Vibrio, and Pseudoalteromonas, are widespread and diverse associates of marine organisms, and despite sharing a (genus) name, contain members that might provide the clues to both the causes and the cures of a range of diseases.
Check out the paper for the full story. |
30 August 2021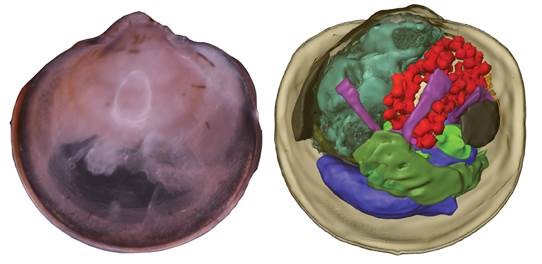 Coming together—symbiont acquisition and early development in deep-sea bathymodioline mussels Citation: Franke M, Geier B, Hammel JU, Dubilier N, Leisch N. (2021). Coming together—symbiont acquisition and early development in deep-sea bathymodioline mussels. Proceedings of the Royal Society B, volume 288, issue 1957; DOI: 10.1098/rspb.2021.1044.▼ Read summaryWe all want to ensure our kids are equipped to survive in the world - provide them with a high-quality education, solid work ethic, self-confidence, but how often do we think about equipping them with the right microbes? Providing offspring with the microbial partners that will ensure proper development and long-term health is a central challenge, but we know very little about how most organisms overcome this challenge because it is so difficult to observe in nature. Understanding symbiont transmission between host generations is particularly challenging in remote locations such as at deep-sea hydrothermal vents and cold seeps, but is important because entire ecosystems in these habitats rely on invertebrates such as Bathymodiolus mussels finding and coming together with particular species of chemosynthetic bacteria that provide them with nutrition. Maximilian Franke and a team of colleagues led by Nikolaus Leisch and Nicole Dubilier from the Max Planck Institute for Marine Microbiology have just provided the most spectacularly detailed insights so far into symbiont transmission in Bathymodiolus mussels. They collected very early developmental stages of several Bathymodiolus species from the Atlantic Ocean and Gulf of Mexico, the smallest of which were symbiont-free pediveliger larvae, less than 400 um in length. The team overlaid a very impressive range of imaging methods including histology, electron microscopy, DOPE-FISH and synchrotron radiation-based micro-computed tomography, to follow the very first stages of symbiont acquisition and development. Symbionts were never present in pediveligers, which is the final free-swimming developmental stage, but were found in all larvae undergoing metamorphosis after settling on the sea floor. Symbionts colonized all epithelial tissues of the developing mussels, in contrast to the adults, where colonization is restricted to the gill epithelia. The range of morphological changes triggered in the animal hosts after symbiont colonization is remarkable, ranging from the cellular level, including loss of cilia and microvilli from the cells colonized by symbionts, to changes in the length and complexity of the digestive tract. Bathymodiolus and its intracellular chemosynthetic symbionts are a beautiful example of coordinated development between animals and microbes that must involve intricate and highly specific mechanisms of inter-domain communication. One interesting finding was the lack of ‘intermediate’ stages between initial colonization by a few symbiont cells, and fully colonized individuals. This could have been missed due to under sampling, however, it may also be that once initiated, the symbiosis rapidly becomes fully established. Even hydrothermal vent mussels seem to have efficient mechanisms to ensure their offspring find the symbiotic bacteria they need for their survival.
|
28 June 2021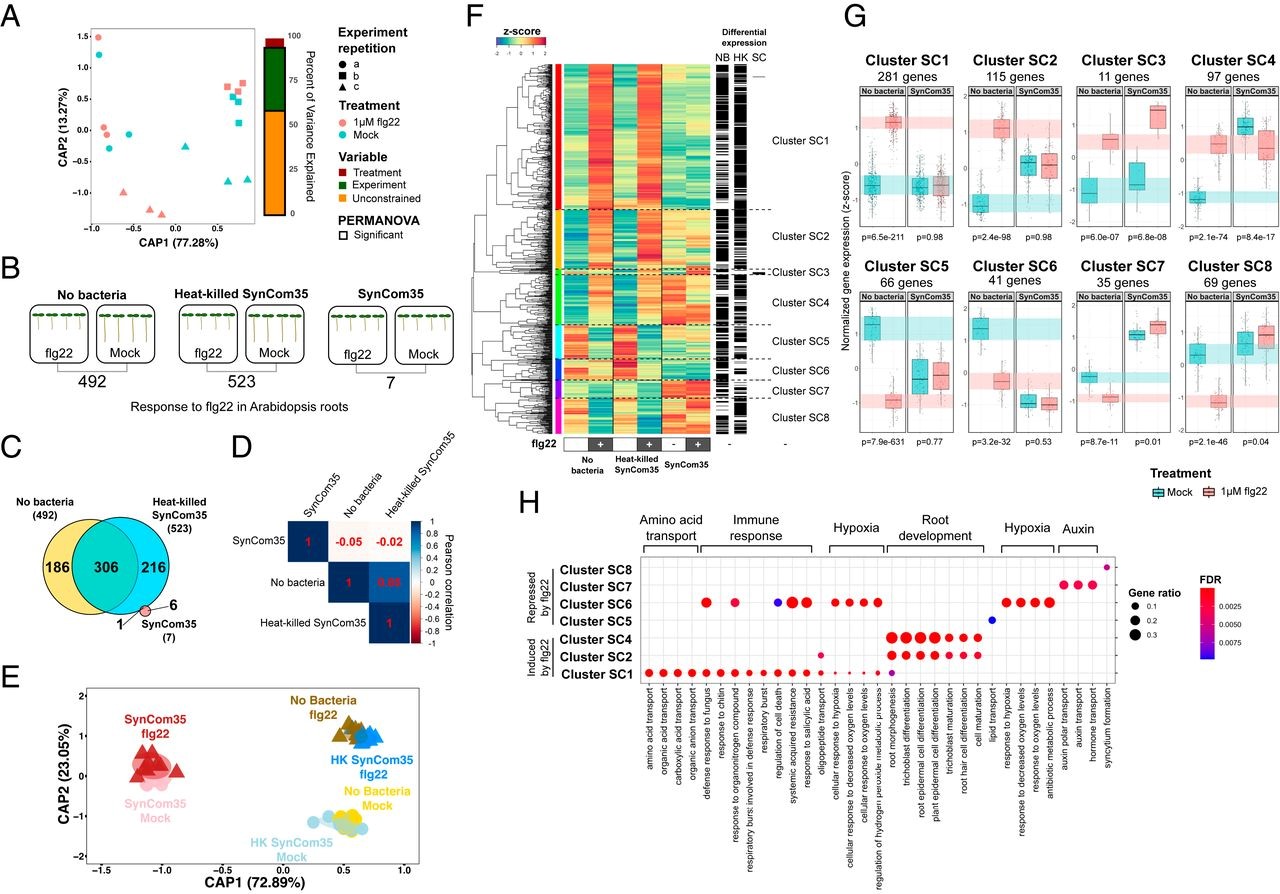 Specific modulation of the root immune system by a community of commensal bacteria Citation: Teixeira PJPL, Colaianni NR, Law, TF, Conway JM, Gilbert S, LiH, Salas-González I, Panda D, Del Risco NM, Finkel OM, Castrillo G, Mieczkowski P, Jones CD, Dangl JL. (2021). Specific modulation of the root immune system by a community of commensal bacteria. Proceedings of the National Academy of Sciences Apr 2021, 118 (16) e2100678118; DOI: 10.1073/pnas.2100678118.▼ Read summaryPlant-associated commensal bacteria represent a subset of those in the bulk soil. This is at least partly due to the sophisticated innate immune system that plants have evolved and allow them to select between beneficial, neutral, or harmful microbes. The MAMP-triggered immunity (MTI) is the first layer of that defence system. It is based on pattern recognition receptors (PRRs) detecting epitopes such as flg22, a 22-amino acids epitope found in the bacterial flagellum. MTI has been thoroughly studied in the context of interaction with pathogens. To interfere with plant innate immunity, pathogens have evolved host-specialised effector proteins often secreted by TSS3 (Type 3 Secretion System). Similar immunomodulation has previously been described by commensal strains but remains poorly understood in the context of community assembly. Teixeira et al. investigated the modulation of Arabidopsis flg22-triggered immune response by a bacterial synthetic community (SynCom) composed of 35 representative commensals. While flg22 treatment induced a transcriptomic response of MTI marker genes both by control plants and plants inoculated with heat-killed bacteria, it was clearly suppressed by the plants inoculated with the living SynCom. Inoculated plants showed a specific transcriptomic pattern and flg22 did not affect assembly community composition. Hence, SymCom assembly actively interferes with flg22-driven MTI. Monoassociations showed that even if MTI suppression is the SynCom dominant trait, taken individually the strains can have inducer, neutral or suppressor effects on the plant response. Ten out of the 35 members of the SynCom were shown to be robust suppressors. Interestingly, the same robust suppressors were also the best root colonisers and were enriched into the endospheric compartment when the non-suppressors were not. Colonisation assays concluded that suppressor strains can enhance root colonisation capacities of other commensals. Thus, strains possessing immunomodulatory activity have a significant role in plant microbiota composition. Finally, the team investigated the mechanisms behind the robust suppression of the plant immune response by Dyella japonica MF79. Whereas its TSS3 was dispensable for the suppression ability, a forward genetic approach identified that a TSS2 (Type 2 Secretion System) was required for the suppression of MTI. No other genes were identified, thus implying that the suppression depends on the extracellular secretion of multiple compounds. These findings suggest that modulation of host immune system is a widespread trait among root-associated bacteria. The presence of suppressor strains also benefiting to the non-suppressors. That low degree of specialisation is hypothesised to be caused by redundant immunosuppressive mechanisms acquired by the different commensals through evolution. Nonetheless, Teixeira et al. yielded highly valuable transcriptomic data that points up a core set of evolutionary conserved defence-related genes that are manipulated by commensal bacteria. These genes are promising candidates as key regulators of the plant immune response to bacteria colonisation and might play a key role in root microbiota homoeostasis.
|
21 June 2021
Microbial hitchhiking: how Streptomyces spores are transported by motile soil bacteria
|
15 June 2021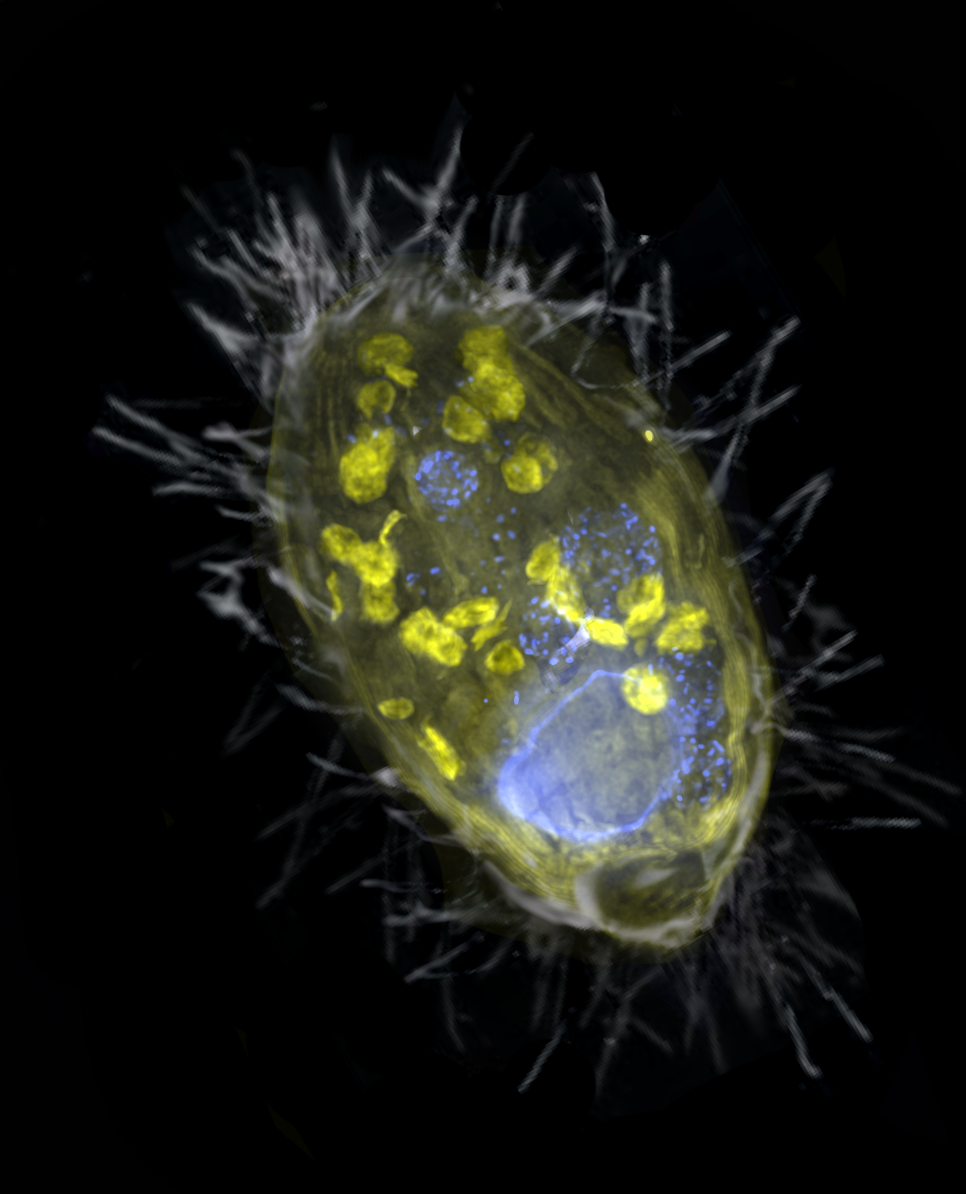
Nitrate is the new oxygen: anaerobic endosymbiont provides energy for its ciliate host through denitrification Citation: Graf JS, Schorn S, Kitzinger K, et al. (2021) Anaerobic endosymbiont generates energy for ciliate host by denitrification. Nature 591: 445–450.▼ Read summary
‘Ca. A. ciliaticola’ belongs to the gammaproteobacteria. Although ‘Ca. A. ciliaticola’ shares a rather similar function with mitochondria, the partnership between the host ciliate and its endosymbiont was established through a separate, more recent endosymbiotic event. Given the great diversity in metabolism and electron acceptors in prokaryotes, this study raises an exciting possibility that there may be other, as-yet undiscovered electron acceptors eukaryotes can use for respiration.

Dr Jana Milucka is a Group Leader in MPI for Marine Microbiology in Bremen. Her research interests span microbiology of methane and nitrogen cycling, anaerobic microbial metabolism and microbial symbioses.
|
31 May 2021Host reproductive cycle influences the pouch microbiota of wild southern hairy-nosed wombats (Lasiorhinus latifrons)Citation: Weiss S, Taggart D, Smith I, Helgen KM, Eisenhofer R. (2021) Host reproductive cycle influences the pouch microbiota of wild southern hairy-nosed wombats (Lasiorhinus latifrons). Animal Microbiome volume 3, Article number: 13 (2021).▼ Read summaryMarsupials are a group of mammals that we share a common ancestor with ~160 million years ago. A key taxonomic feature of marsupials is their early birth (1-6 weeks) followed by further development of the joey (young) in a pouch. See this cool video showcasing the epic journey joeys must make to reach their safety of their mother’s pouch. Being born young and without an immune system in a microbially rich world is challenging, and some known mechanisms for protecting the vulnerable joey include the transfer of antibodies and immune cells through milk, and the secretion of antimicrobial peptides into the pouch. However, little is known about the microbiology of the pouch – are there microbes present, and if so, are they influenced by the host’s reproductive status? To fill this knowledge gap, we caught and sampled the pouch and other body sites from 26 wild female southern hairy-nosed wombats in South Australia (including both reproductively active females and subadults). We determined that there were indeed microbes present in the wombat pouch by using qPCR of the 16S rRNA gene and comparing 16S rRNA gene abundance to negative controls (sampling and extraction blanks). Next, using 16S rRNA gene sequencing we found that microbial composition of reproductively mature wombat pouches was distinct to that of other sample types, including subadult pouch samples, which resembled skin samples. Additionally, the number of amplicon sequence variants (ASVs) was drastically lower in reproductively active female pouches (mean 19) compared to subadult pouches (mean 941). Five microbial genera accounted for >90% of the microbial community in reproductively active pouches, and three of the five most abundant ASVs in these samples had closest BLAST matches to 16S rRNA gene sequences previously isolated from tammar wallaby pouches (another marsupial species). Overall, our findings indicate that there appears to be a high degree of host filtering for what types of microbes can live in the pouch of reproductively active wombats. For the microbes that can survive in the pouch, how they do so, and what functions they possess remain to be discovered. The similarity of some of these wombat pouch microbes to microbes found in another marsupial species could indicate some degree co-speciation between host and pouch microbes. One possibility is that these pouch microbes could be beneficial to the host by occupying space and deterring pathogenic microbes from taking residence in the pouch. There is much still to learn about how microbes influence the evolution and ecology of Australia’s unique marsupials. |
14 May 2021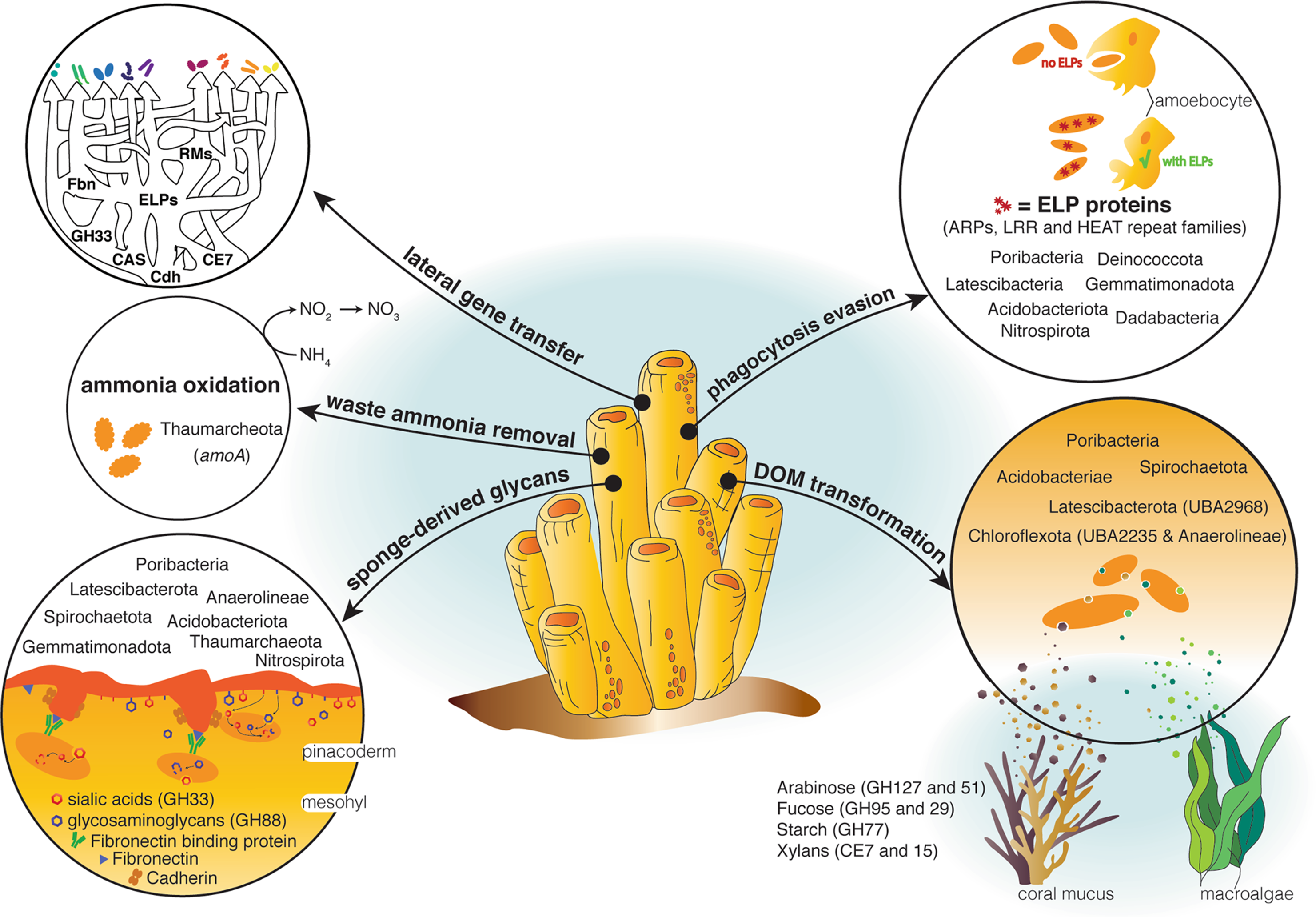 A genomic view of the microbiome of coral reef demosponges Citation: Robbins SJ, Song W, Engelberts JP et al. A genomic view of the microbiome of coral reef demosponges. ISME J (2021). doi.org/10.1038/s41396-020-00876-9.▼ Read summaryMarine sponges are important for the health of coral reefs because of their role in the so-called “Sponge-Loop.” The crux of the Sponge Loop is that while coral reefs are one of the most productive ecosystems on earth, they are often referred to as “marine deserts” because ambient nutrient concentrations are low. Nutrient retention, then, is critical. Sponges filter out large quantities of organic matter released into the water column by other organisms living on the reef and convert it into sponge tissue. Their cells are then shed into the water column where they can be eaten by detritovores and the organic matter recycled back into the food chain, keeping these nutrients locked within the reef. Tropical sponges, like most animals, host a variety of microbial symbionts (bacteria and archaea). These symbionts can make up to ~35% of the sponge’s total biomass and are thought to carry out a range of processes that are important for maintaining sponge health. For example, sponge symbionts are thought to provide the sponge with vitamins, remove waste ammonia, and potentially participate in the sponge loop by transforming components of dissolved organic matter. However, the majority of symbiont lineages found in tropical marine sponges remain largely uncharacterised, hampering our ability to identify core features of sponge symbionts, those that underpin the relationship between the host and its microbes. To address this, we undertook an analysis of ~1200 metagenome-assembled genomes (MAGs) derived from seven marine sponge species and spanning 25 microbial phyla, allowing us to map functions thought to be important for sponge-microbe symbiosis across the vast majority of microbial taxa commonly found in marine sponges. This allowed us to identify specific functional guilds for the transformation of sugars common in coral reef dissolve organic matter, suggesting that microbes play a role in the Sponge Loop. We also show that the Thaumarchaeota are a keystone taxon for ammonia oxidation, and that many genes thought to be critical for maintaining sponge symbiosis (e.g. euk-repeat proteins) are not universally distributed among sponge symbionts, requiring some as-yet unknown mechanism in these lineages. Finally, we provide evidence that many of the genes thought to underpin sponge-microbe symbiosis, like the glycosyls hydrolases for transforming reef DOM, eukaryote-like proteins for evading the host immune system, and CRISPR and restriction-modification system genes for fending off viral infection, have been laterally transferred between disparate microbial lineages. Taken together, these data illustrate how evolutionary processes have partitioned ecological functions across sponge symbiont lineages, allowing them to occupy or share specific niches and live symbiotically with their sponge hosts. |
14 May 2021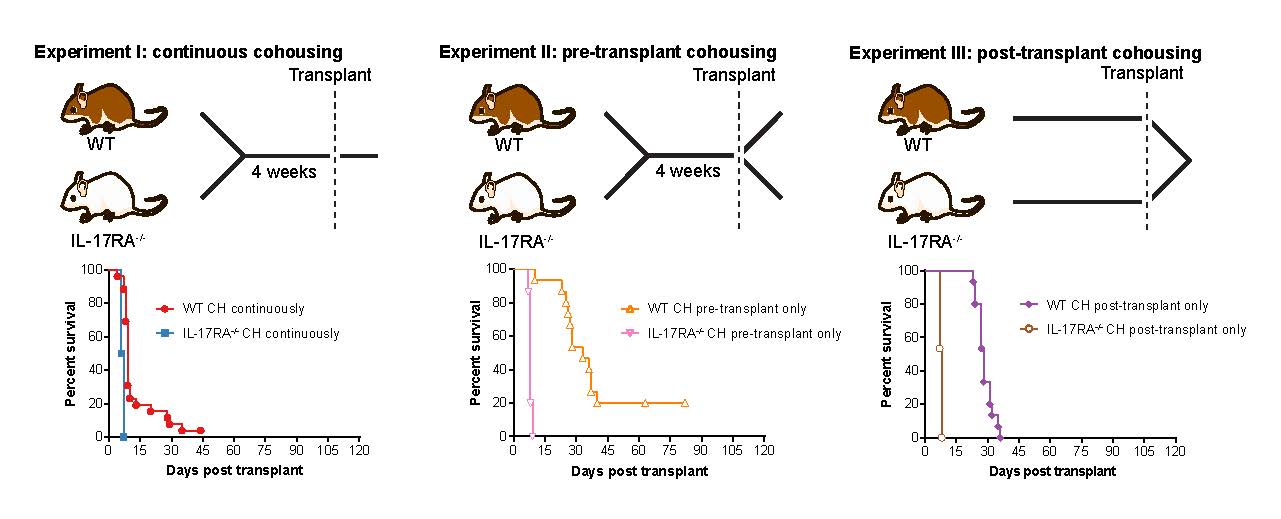 Continuous pre- and post-transplant exposure to a disease-associated gut microbiome promotes hyper-acute graft-versus-host disease in wild-type mice Citation: Bowerman KL, Varelias A, Lachner N, Kuns RD, Hill GR, Hugenholtz P. (2020) Continuous pre- and post-transplant exposure to a disease-associated gut microbiome promotes hyper-acute graft-versus-host disease in wild-type mice. Gut Microbes, 1-17. doi.org/10.1080/19490976.2019.1705729.▼ Read summaryGraft-versus-host disease (GVHD) is a serious complication of hematopoietic stem cell transplantation occurring in 30-50% of cases. The gut microbiome has been observed to fluctuate both before and during the development of GVHD, creating a need to investigate whether there is a critical period during which the gut community composition can critically influence disease development. To investigate the contribution of the gut microbiome at different timepoints around the induction of GVHD, a team from the University of Queensland and QIMR Berghofer Medical Research Institute employed a mouse model system involving cohousing different strains of mice, with known divergent gut microbiomes, before and after stem cell transplant. They found that priming wild-type mice by cohousing them with a highly GVHD susceptible mouse strain prior to transplant did not affect disease progression, however, if cohoused after transplant, accelerated disease was observed. When wild-type mice were cohoused both before and after transplant, the effect of priming and post-transplant exacerbation were additive, resulting in a greater acceleration of disease beyond that seen with cohousing after transplant alone. Metagenomic analysis of the microbiome revealed pre-transplant cohousing was associated with the transfer of specific species within two as-yet-uncultured genera of the bacterial family Muribaculaceae; CAG-485 and CAG-873. Post-transplant, the authors observed GVHD-associated blooms of Enterobacteriaceae members Escherichia coli and Enterobacter hormaechei subsp. steigerwaltii, and a hyper-acute GVHD gut microbiome distinct from that associated with delayed-onset disease (>10 days post-transplant). These results clarify the importance of the peri-transplant microbiome in the susceptibility to acute GVHD post-transplant and demonstrate the species-specific nature of this association. |
26 April 2021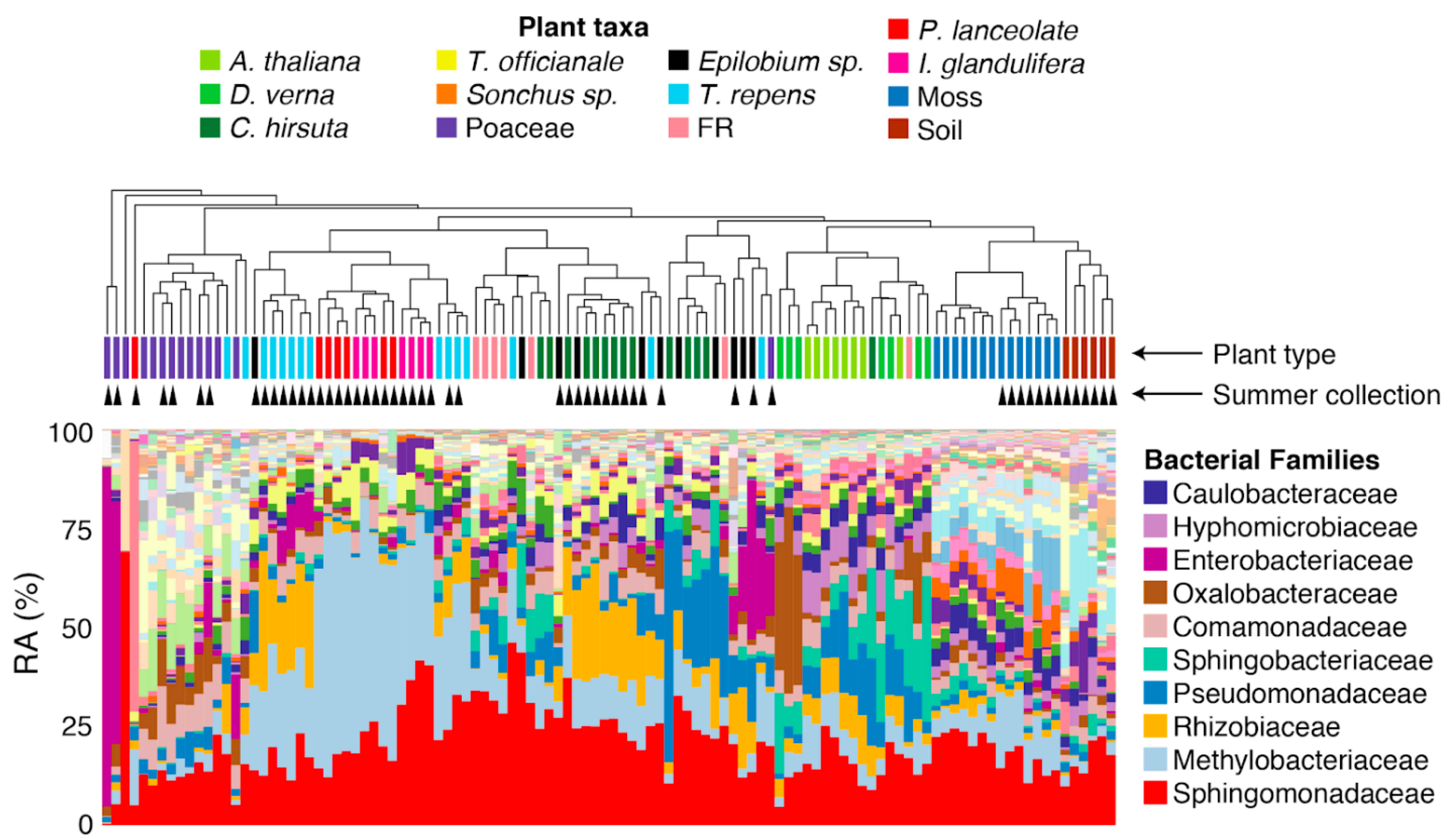 Contrasting patterns of microbial dominance in the Arabidopsis thaliana phyllosphere Citation: Lundberg DS, de Pedro Jové R, Pramoj Na Ayutthaya P, Karasov TL, Shalev O, Poersch K, Ding W, Bollmann-Giolai A, Bezrukov I, and Weigel, D. Contrasting Patterns of Microbial Dominance in the Arabidopsis Thaliana Phyllosphere. BioRxiv https://doi.org/10.1101/2021.04.06.438366.▼ Read summaryAboveground organs of plants, or the phyllosphere, represent open and fluctuating habitats for microbes originating from various sources, such as soil, seeds, and from neighboring plants transferred via air, rain, insects, or other means. To date, the strain-level understanding of phyllosphere microbiome membership is limited, and best studied in the genus Pseudomonas. To better understand the ecology and host specificity in the phyllosphere microbiome, Lundberg et al. turned to another abundant yet less understood genus, Sphingomonas. The authors employed genomic techniques to analyze leaves of wild Arabidopsis thaliana and neighboring other plant species in spring, when A. thaliana plants were flourishing, and late summer, when there was no living A. thaliana. Amplicon sequencing of 16S rRNA genes revealed that both Sphingomonas and Pseudomonas dominated the leaf microbiome and were common endophytically, but Sphingomonas colonized more consistently in different seasons and had a broader host range than Pseudomonas. The authors established a genome-sequenced culture collection of endophytic Sphingomonas, which allowed for in-depth genome analyses at the strain level. The analysis revealed that Sphingomonas genomes are more diverse than Pseudomonas, and 16S rRNA information falls short in comprehending the diversity of plant-associated Sphingomonas. The authors also devised a cost-efficient metagenome approach in which Sphingomonas or Pseudomonas were enriched using cultivation on selective media before sequencing. The metagenome data showed that the most abundant strains of Sphingomonas and Pseudomonas in plants were not well represented in the soil, whereas some strains were shared between different plant species and, in some cases, between different seasons. These results suggest that leaf-to-leaf transmission may be a major source of these strains to the next generation. Overall, this study demonstrates the importance of strain-level analyses of the plant microbiome, and reveals shared and contrasting modes of interactions between leaves and dominant bacterial species in the phyllosphere. |
22 April 2021
Experimental evidence of microbial inheritance in plants and transmission routes from seed to phyllosphere and root 
Citation: Abdelfattah A, Wisniewski M, Schena L, Tack AJM.(2021). Experimental evidence of microbial inheritance in plants and transmission routes from seed to phyllosphere and root. Environmental Microbiology. doi.org/10.1111/1462-2920.15392.▼ Read summarySoil is widely considered as the origin and main source of microorganisms associated with plants. However, the potential contribution of seeds to the assembly of the plant microbiome cannot be ignored. Seed microbiota remained relatively unknown, and were much less studied compared to well characterized plant compartments such as rhizosphere and phyllosphere. Studies that demonstrate the microbiome's vertical inheritance (from seed to seedling) under natural conditions are challenging once plants are exposed to the soil with its high microbial diversity. To overcome this problem, Abdelfattah and colleagues developed a microcosm to grow common oak seedlings in microbe-free conditions while keeping the below- and aboveground plant parts separated. Using an amplicon-based approach of the ITS and 16S regions to characterize the fungal and bacterial community present in the embryo and pericarp of acorns, roots and phyllosphere of the developing seedling, the author were able to identify the transient and vertically transmitted microbiota from the seeds. The authors found that the composition of the microbial community in embryo and pericarp greatly differed and the major part of the inherited microbiome is transmitted from the seed to the seedling. This emphasizes the ecological role of seeds as a reservoir and source for community assembly in new seedlings. Interestingly, the phyllosphere microbiota was derived mainly from the embryo microbiome, while the root microbiome represented a distinct subset of the seed microbiome. The authors also discuss the lack of information about the microbial community from embryo, since several unidentified fungal and bacterial taxa were found. Since the authors demonstrated that this seed environment is a crucial part of the vertical inheritance process, the importance of discovering of novel species that occupy this niche becomes apparent. In summary, this study provides new information on the source and transmission of plant-associated microbes. This knowledge could increase our understanding of plant biology, and help in future biotechnological applications such as new strategies for breeding healthier crops.
|
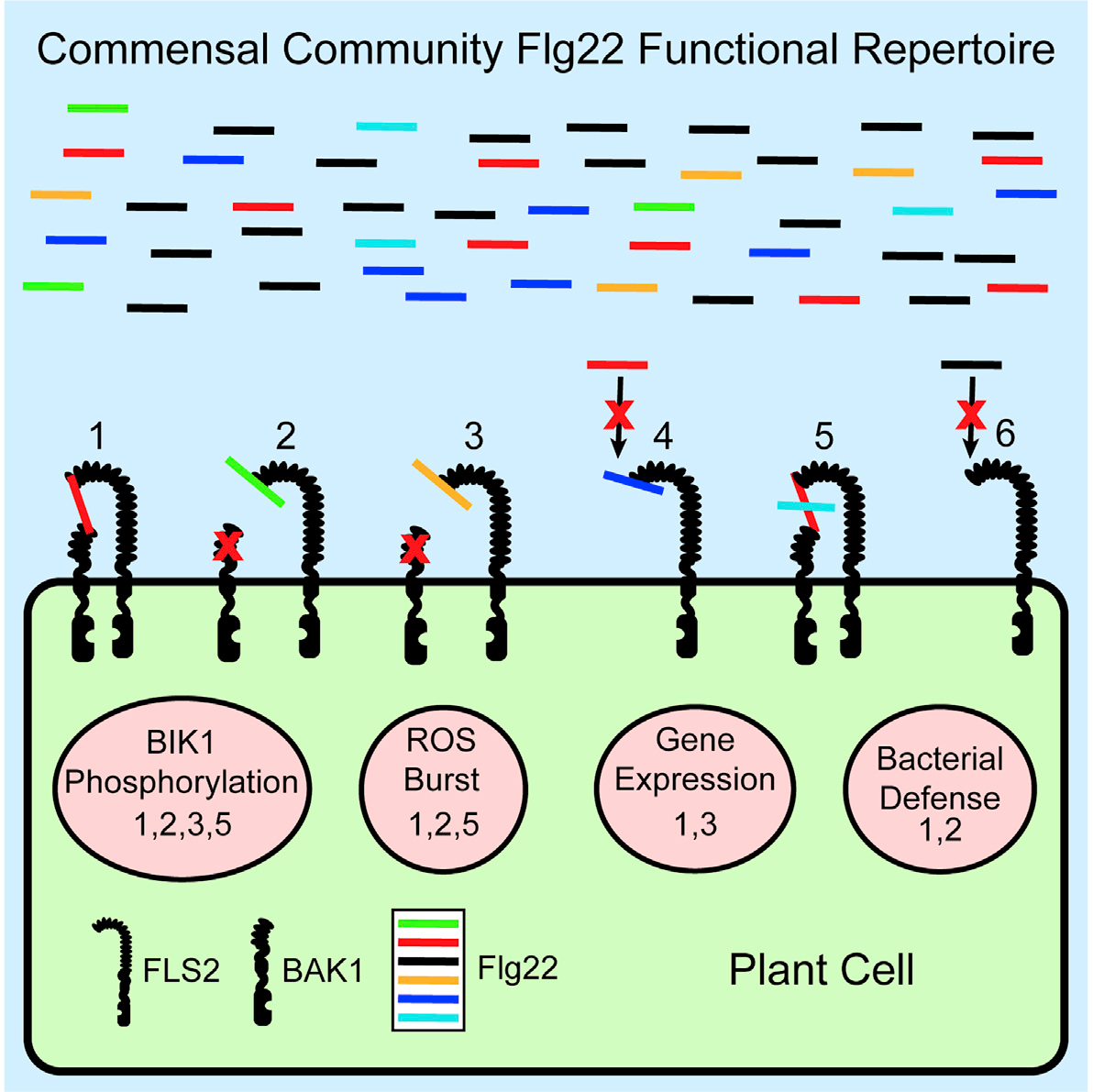
14 April 2021A complex immune response to flagellin epitope variation in commensal communities Citation: Colaianni NR, Parys K, Lee H-S, Conway JM, Kim NH, Edelbacher N, Mucyn TS, Madalinski M, Law TF, Jones CD, et al., 2021. A complex immune response to flagellin epitope variation in commensal communities. Cell Host Microbe. doi: 10.1016/j.chom.2021.02.006.▼ Read summaryIn nature, plants interact with many different microbes (pathogenic, beneficial or commensal). All these microbes produce similar conserved molecules known as microbe-associated molecular patterns (MAMPs). Perception of MAMPs by specialized plant receptors, known as pattern recognition receptors or PRRs, can lead to MAMP-triggered immunity (MTI). MTI is a first layer of defense employed by plants to restrict microbial colonization and pathogenesis. One well-characterized MAMP-PRR pair is that of bacterial flagellin and FLAGELLIN SENSING 2 (FLS2). Flagellin is the building block of bacterial flagellum, required for bacterial motility, and contains flg22, a 22-amino-acid peptide known for its immune-eliciting potential. Despite the fact that different bacteria are motile due to flagellin and possess flg22 peptides, most plant niches are colonized by millions of bacteria. Therefore, a delicate interplay occurs between flg22 of plant-associated bacteria and the plant immune system. Researchers from Dangl and Belkhadir groups aimed to characterize the flg22 repertoire of Arabidopsis commensal bacteria and their ability to induce MTI and inhibit plant growth. Genome mining in more than 600 bacterial isolates revealed their categorization in three clades, based on diversity of flagellum proteins and flg22 sequence similarity. Synthesis of 97 flg22 peptide variants (representative of the three clades) and subsequent screening for MTI, revealed that some are immunogenic (mostly from Clade I bacteria; β-/γ-Proteobacteria) while most of the peptides (mostly from Clade III bacteria; Rhizobiales, Caulobacterales) avoid to induce MTI or cause growth inhibition via different modes of action. Based on their modes of action flg22 peptides were categorized as evading (avoid MTI activation), deviant (different levels of MTI) and antagonistic (antagonizing MTI activation by immunogenic peptides). Further analysis revealed that prevalent members of rhizosphere and phyllosphere bacterial communities produce antagonistic and evading flg22 peptides, achieving as such low MTI activation and successful colonization. Analysis of a complex synthetic microbial community colonizing roots and leaves of Arabidopsis in vitro, revealed that evading flg22 peptides are enriched in both root- and shoot-associated microbes, while the immunogenic ones are depleted. However, under conditions of salt stress, leading to a weakened plant immune system, there is enrichment of immunogenic flg22 variants in Arabidopsis-associated microbes. This finding suggests that optimal function of the immune system is required to monitor bacterial presence based on flg22 immunogenicity. This study demonstrates that complex plant-associated bacterial communities produce a cocktail of flg22 peptides to avoid strong MTI activation and represents an important step towards understanding why some microbes are successful plant colonizers while others are not. Katarzyna Parys is a collaborator at the Gregor Mendel Institute of Molecular Plant Biology GmbH, in Vienna, Austria.
|
26 March 2021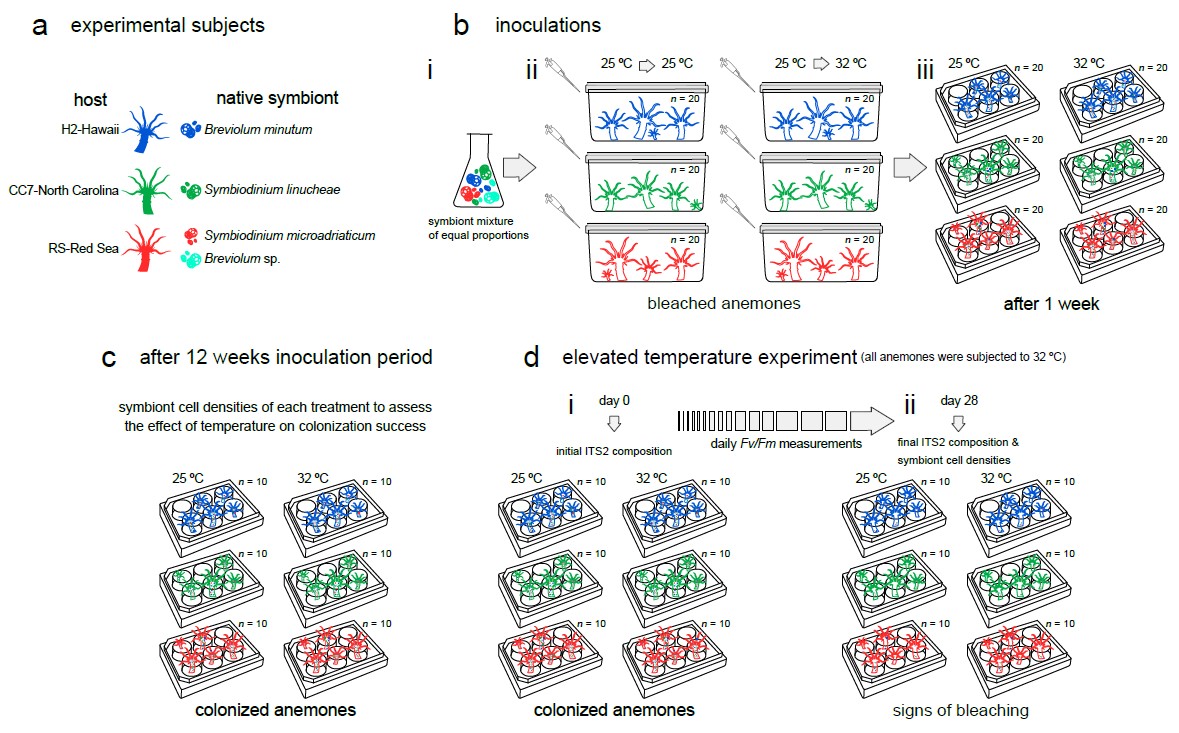 Temperature transcends partner specificity in the symbiosis establishment of a cnidarian Citation: Herrera M, Klein SG, Campana S, et al. Temperature transcends partner specificity in the symbiosis establishment of a cnidarian. ISME J 15, 141–153 (2021). doi.org/10.1038/s41396-020-00768-y.▼ Read summaryCorals live in a delicate symbiotic relationship with dinoflagellate algae in the family Symbiodiniaceae. This relationship, however, is fragile and can break down in response to environmental stressors such as increased temperature, a process known as coral bleaching. As warming sea surface temperatures, consequence of climate change, become more frequent and severe, coral reef ecosystems continue to decline worldwide. Yet, contrary to increasing research efforts on coral bleaching, little is known about the establishment and development of novel symbioses in rapidly warming environments. Certainly, symbiotic flexibility is an important factor when predicting how corals might respond to increasing temperatures. Unlike other studies, Herrera et al. (From the Red Sea Research Center in KAUST/Saudi Arabia) tested different combinations of host genotypes and their native symbionts, thus taking into account genotypic variability among hosts but also the potential constraints of symbiosis and functional diversity (e.g., thermal sensitivity) within Symbiodiniaceae. The authors used the flexible model system Exaiptasia pallida to investigate how host specificity and temperature affect the symbiosis establishment of a cnidarian. They inoculated symbiont-free anemones originating from three geographically distinct locations (Hawaii, North Carolina, and the Red Sea) using a mixture of native symbiont strains (Breviolum minutum, Symbiodinium linucheae, S. microadriaticum, and a Breviolum ecotype from the Red Sea) at ‘optimal’ and elevated temperatures and performed deep ITS2 sequencing to assess colonization dynamics across the different thermal conditions. Furthermore, they measured PSII photochemical efficiency (Fv/Fm) as a proxy of the physiological performance of symbioses over time which highlights the importance of heterotrophic feeding on the response to temperature. This study highlights the role of temperature and partner fidelity in the establishment and performance of symbiosis and further adds to the body of work showing that pre-exposure to elevated temperature is crucial for the thermal resilience of the cnidarian holobiont. Moreover, it shows the potential of using species from the Red Sea to study thermal resilience of the cnidarian holobiont. Finally, the authors also demonstrate the importance of active feeding for enduring thermal stress. |
7 March 2021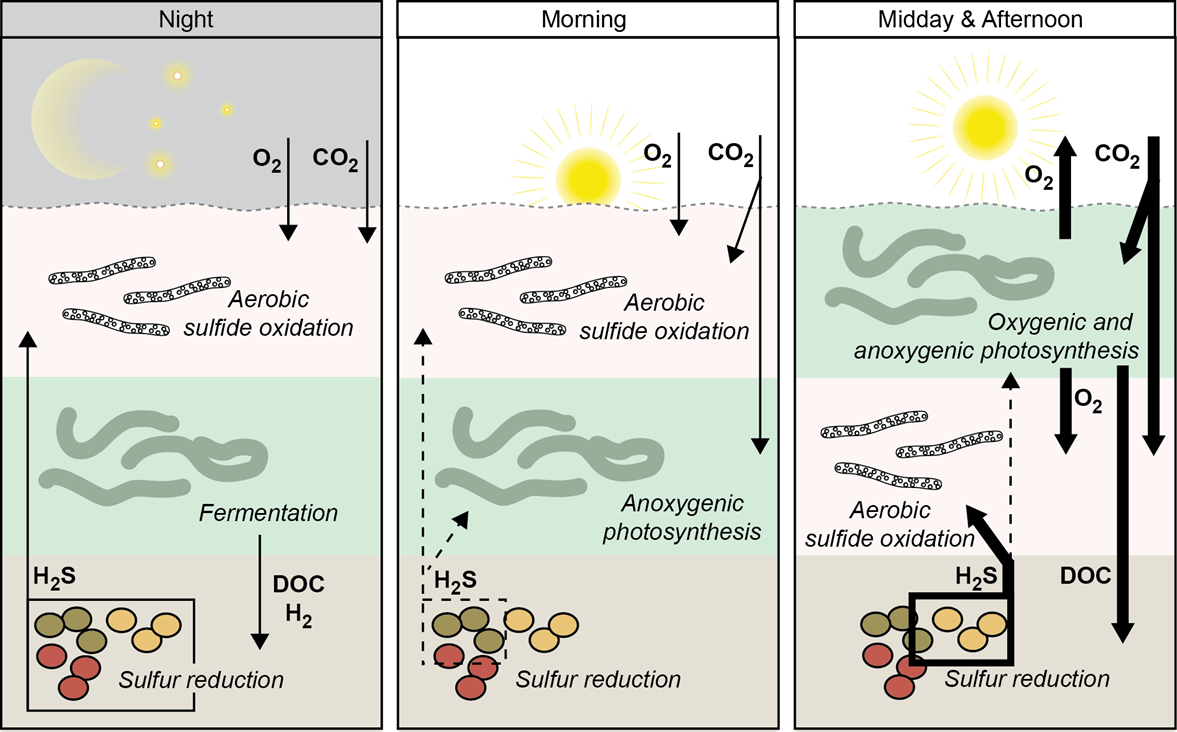 Versatile cyanobacteria control the timing and extent of sulfide production in a Proterozoic analog microbial mat Citation: Klatt J M, Gomez-Saez GV, Meyer S, Ristova PP, Yilmaz P, Granitsiotis MS, et al. (2020). Versatile cyanobacteria control the timing and extent of sulfide production in a Proterozoic analog microbial mat. The ISME Journal. doi:10.1038/s41396-020-0734-z.▼ Read summaryCyanobacterial mats were hotspots of biogeochemical cycling throughout the Proterozoic. |
7 March 2021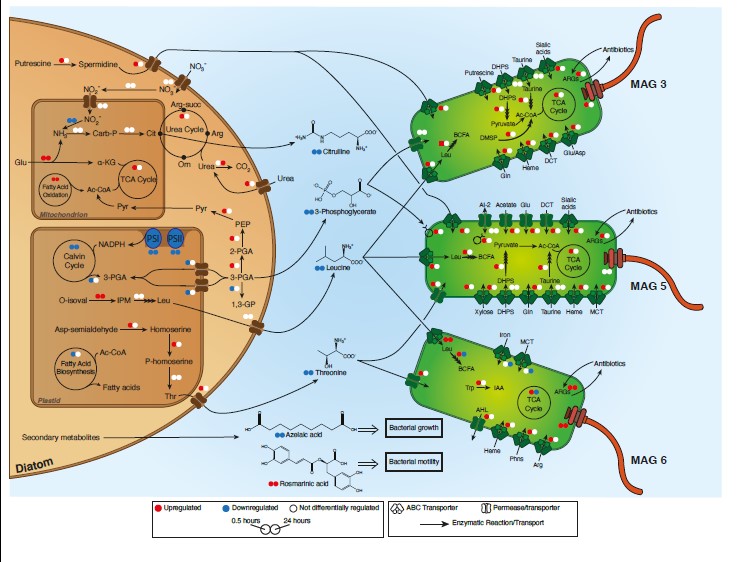 Diatom modulation of select bacteria through use of two unique secondary metabolites Citation: Shibl AA, Isaac A, Ochsenkühn MA, Cárdenas A, Fei C, Behringer G, Arnoux M, Drou N, Santos MP, Gunsalus KC, Voolstra CR, Amin SA. Diatom modulation of select bacteria through use of two unique secondary metabolites. Proc Natl Acad Sci U S A. 2020 Nov 3;117(44):27445-27455.▼ Read summaryMulticellular eukaryotes have dedicated structures to house microbiomes, whereas the mechanisms that enable diatoms and other phytoplankton species to actively modulate incoming microbes are mostly unknown. The marine microbial ecology laboratory team at the New York University in Abu-Dhabi and their colleagues from the MPI in Bremen, as well as scientists from KAUST and the University of Konstanz explored the ability of a widespread diatom, Asterionellopsis glacialis, to adopt specific mechanisms to promote association with potentially beneficial symbionts while repelling opportunists. They employed an integrated multi-omics approach (genomics, transcriptomics, metabolomics) to tease apart the functional and metabolic capacities of a bacterial consortium and a phytoplankton within a host-microbiome system. In addition, they targeted two bacterial genera, Roseobacteria and Alteromonas, to test the effects of two enigmatic secondary metabolites, azelaic acid and rosmarinic acid, on the growth and behavior of associated bacteria within the diatom photosphere. Their results suggest that a host phytoplankton cell goes through major transcriptional and metabolic reprogramming in order to modulate its microbiome through secondary metabolites. Therefore, the innate ability of an important unicellular eukaryotic group to modulate select bacteria in their microbial consortia is similar to higher eukaryotes, using unique secondary metabolites that regulate bacterial growth and behavior inversely across different bacterial populations. |
 About the second author: Prof. Olubukola Oluranti Babalola is a Principal Investigator, a Microbial Biotechnology Research team. Her laboratory is a mini United Nations, with students from within and outside Africa, making a substantial contribution to Science, her original research motivation. Her wealth of international experience spans the Americas, Asia, Europe, & Oceania. She enjoys international collaborations, research grants, gender lens applications & many awards, including being proclaimed the finalist, GenderInSite 2020.
About the second author: Prof. Olubukola Oluranti Babalola is a Principal Investigator, a Microbial Biotechnology Research team. Her laboratory is a mini United Nations, with students from within and outside Africa, making a substantial contribution to Science, her original research motivation. Her wealth of international experience spans the Americas, Asia, Europe, & Oceania. She enjoys international collaborations, research grants, gender lens applications & many awards, including being proclaimed the finalist, GenderInSite 2020. About the author:
About the author:  About the author
About the author  About the author:
About the author:  About the first author:
About the first author:  About the authors:
About the authors: 
 About the first author:
About the first author: 
 About the authors: Dr
About the authors: Dr  Dr
Dr 
 About the first author:
About the first author:  About the first author:
About the first author: 
 About the first author :
About the first author :  About the co-authors :
About the co-authors : 
 About the first author and the group:
About the first author and the group:  About the first author:
About the first author:  About the first author:
About the first author: Also view the ECSC's other activities: ISME Conversations